Introduction
Cellular senescence is an irreversible growth arrest that can occur after repeated cell division (replicative senescence). It can also be prematurely induced by stress conditions such as oxidative stress, DNA damage and mitogenic stress (stress-induced senescence) [123]. Senescent cells cannot divide even if stimulated by mitogens, but they are not physiologically inert displaying altered phenotypic changes and gene expression. Furthermore, they secrete degradative enzymes, inflammatory cytokines, and growth factors, leading to a shift from extracellular matrix synthesis to degradation. Therefore, senescence is considered as a major factor contributing to age- or disease-associated tissue dysfunction and pathology. Replicative senescence is mediated by the p53-p21-pRB pathway; whereas stress-induced senescence is mediated by the p16-pRB pathway [456]. Senescent cells are characterized by telomere shortening and decreased telomerase activity. In addition, senescence-associated-beta-galactosidase (SA-β-Gal) activity is used to identify the occurrence of senescence [789].
Diabetes mellitus (DM) is a major public health problem worldwide. The number of DM patients is projected to reach 300 million in 2025 (International Diabetes Federation, 2001) [10]. Untreated DM can cause many complications, such as cardiovascular disease, chronic renal failure, retinopathy and neuropathy [11]. DM is also considered an important etiological factor in disc degeneration [121314]. Previous studies have reported a higher incidence of degenerative disc diseases in patients with DM than in the patients without DM [15]. A considerable portion of patients who undergo spine surgery suffer from DM. Their surgical outcome was reported to be poor, as compared to non-DM patients [1617]. Recently, there have been reports on the association of senescence and disc degeneration [181920212223]. Premature disc degeneration is accompanied by the loss and/or dysfunction of notochordal cells. However, senescence of notochordal cells as a biological mechanism and the role of senescence in disc degeneration are not well understood to date.
The glucose-mediated increase in oxidative stress, such as the reactive oxygen species (ROS), is a main biochemical pathway in diseases associated with DM [242526]. The mitochondria is the main source of endogenous ROS in most mammalian cell types [27]. ROS play a role in various cellular processes, such as senescence, which appear to cause cellular damage and physiological dysfunction. Therefore, the accumulation of ROS is reportedly associated with a variety of diseases including neurodegenerative diseases, DM, cancer, premature aging, and inflammatory disorders [2829]. To date, little information is available about the effect of DM on the senescence of notochordal cells and premature disc degeneration. It is important to clarify the effect of DM on the senescence of notochordal cells to prevent or to delay premature disc degeneration in young patients with DM. We therefore investigated the effect of DM on mitochondrial damage, oxidative stress, senescence of nucleus pulposus (NP) cells, and premature disc degeneration.
Materials and Methods
1. Rat notochordal cells culture and high glucose treatments
All lumbar intervertebral discs (L1-6) were harvested from 4-week-old male Sprague Dawley rats (Orient Bio., Seoul, Korea) immediately after sacrifice. The discs were carefully dissected under a microscope to obtain only the gelatinous NP tissue, and the harvested NP tissue was pooled in α-minimum essential medium (α-MEM, Gibco BRL, Grand Island, NY, USA). The cells were released from the NP tissue in Hank's Balanced Salt Solution (Hyclone, Ottawa, ON, Canada) with 0.02% pronase (Sigma-Aldrich, St. Louis, MO, USA) by vigorous pipetting. After the cells grew to confluence, they were split once (passage 1) and allowed to reach confluence again. When the cells reached 80%-90% confluence they were placed in either 10% fetal bovine serum (FBS; normal control) or 10% FBS plus two different high glucose concentrations (0.1 M and 0.2 M, experimental conditions) for 1 and 3 days. The study was approved by the Institution's Animal Care and Use Committee.
2. Detection of mitochondrial damage
Mitochondrial damage of notochordal cells was detected with the mitochondrial transmembrane potential apoptosis detection kit (Abcam Plc, Cambridge, UK). The kit utilizes MitoCapture a cationic dye that differentially fluoresces in healthy and damaged apoptotic cells. In damaged apoptotic cells, MitoCapture cannot aggregate in the mitochondria due to the altered mitochondrial transmembrane potential, and thus it remains in the cytoplasm in its monomeric form, fluorescent green. The cells were seeded on a glass cover slip in 12-well culture plates and then incubated with the MitoCapture reagent in a prewarmed incubation buffer in a humidified atmosphere for 30 minutes at 37℃. Fluorescent signals were detected with a fluorescence microscope (Image-Pro Plus, Media Cybernetics, Rockville, MD, USA; Olympus CO., Tokyo, Japan).
3. Intracellular ROS measurement
Intracellular ROS accumulation was measured by ROS detection with H2DCF-DA (Sigma-Aldrich) according to the manufacturer's instructions. Notochordal cells were stained with 25 µM of H2DCF-DA in phosphate buffered saline (PBS) in a humidified atmosphere (95% air, 5% CO2) for 30 minutes at 37℃. Fluorescence intensity was measured with BD FACS Canto flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
4. Expression of manganese superoxide dismutase (MnSOD)
Notochordal cells were incubated with rabbit anti-MnSOD (Millipore Co., Millipore Billerica, MA, USA) anti-body in a blocking solution for 1 hour at room temperature. After washing, fluorescence-conjugated Cy3-Donkey anti-rabbit antibody (Millipore Co.) and Hoechst Nuclear HCS solution (Millipore Co.) in blocking solution were added and the cells were incubated for 1 hour in the dark. After further washing steps, characteristic markers of organelle phenotype were determined by fluorescence microscopy (Image-Pro Plus, Olympus).
5. Expression of catalase
The intracellular production of catalase by notochordal cells was measured with the Catalase Assay Kit (Cayman Chemical Co., Ann Arbor, MI, USA) according to the manufacturer's instructions. The cells were sonicated on ice in 50 mM potassium phosphate, pH 7.0 containing 1 mM ethylenediaminetetraacetic acid. Absorbance was measured at 540 nm using a plate reader (Molecular Devices, Sunnyvale, CA, USA).
6. Determination of senescence
The SA-β-Gal activity in the notochordal cells was determined using a SA-β-Gal staining kit (Cell Signaling Technology). The cells were plated on to poly (L-lysine)-coated 12-mm glass cover slips and fixed with 2% formaldehyde and 0.2% glutaraldehyde in PBS for 15 minutes at room temperature. The slides were rinsed with PBS and then incubated for 8 hours at 37℃ with fresh SA-β-Gal staining solution containing 40 mM citric acid/sodium phosphate (pH 6.0), 150 mM NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mg/mL of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). After rinsing with 70% glycerol, blue-stained cells were identified as senescent cells under a microscope. All notochordal cells and SA-β-Gal-positive notochordal cells on the entire section were counted (×200) and the percentage of SA-β-Gal-positive notochordal cells was calculated.
7. Determination of telomerase activity
The telomerase activity was analyzed using a TeloTAGGG Telomerase PCR ELISA Plus Kit (Roche, Mannheim, Germany), according to the manufacturer's instructions. Using the ELISA method, the amplified products were immobilized on streptavidin-coated microtiter plates via biotin-streptavidin interaction. Thereafter the amplifications were detected by anti-digoxigenin antibodies conjugated to peroxidase. After addition of the peroxidase substrate (3, 3', 5, 5'-tetramethylbenzidine), the amount of TRAP products was determined by measurement of absorbance at 450 nm using a microplate reader.
8. Expressions of p53, p21, pRB, and p16
Notochordal cells were plated on to the poly (L-lysin)-coated 12 mm glass cover slips. The cells were fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature, and washed with PBS (pH 7.4). The cells were permeabilized with 0.2% TritonX-100 in PBS for 10 minutes. After incubation for 30 minutes at room temperature with PBS containing 3% BSA (blocking solution), cells were incubated at 4℃ overnight with the primary antibody (1:100 dilution) for p53 (Novus Biologicals, Littleton, CO, USA), p21 (Abcam Plc, Cambridge, UK), pRB (Biorbyt Ltd., Cambridge, UK), and p16 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, the anti-rabbit Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) and anti-mouse Alexa Fluor 555 (Invitrogen) fluorescence-conjugated IgG antibodies (1:5,000 dilution) in the blocking solution were added and incubated for 1 hour at room temperature in the dark. The cover slips were mounted on to the objective glasses with mounting medium containing 4, 6-diamidino-2-phenylindole (Vector, Burlingame, CA, USA). Fluorescent signals were detected by fluorescence microscopy (Olympus CO.).
Results
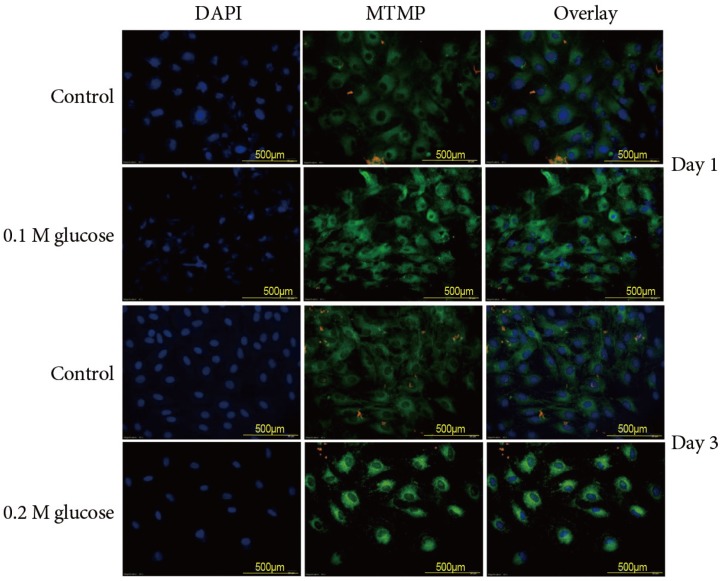
1. Excessive generation of ROS through mitochondrial damage
Immunofluorescence of rat notochordal cells treated with the two high glucose concentrations for 1 and 3 days demonstrated an enhanced disruption of the mitochondrial transmembrane potential (green color), which indicated mitochondrial damage, as compared to the control (Fig. 1). The expression of the mitochondrial transmembrane potential (green color) was detected in the nuclei of notochordal cells, whereas the expression of the mitochondrial transmembrane potential was observed in not only the nuclei of notochordal cells but also in the cytoplasm around the nuclei. Flow cytometry showed that 0.1 M and 0.2 M glucose enhanced ROS generation (% total) in rat notochordal cells at 1 day (18.7% vs. 23.5%, p<0.05; 18.7% vs. 37.4%, p<0.01), as compared to the normal control. Flow cytometry showed that 0.1 M and 0.2 M glucose enhanced ROS generation (% total) in rat NP cells at 3 days (30.8% vs. 39.1%, p<0.05; 30.8% vs. 42.8%, p<0.05), as compared to the normal control.
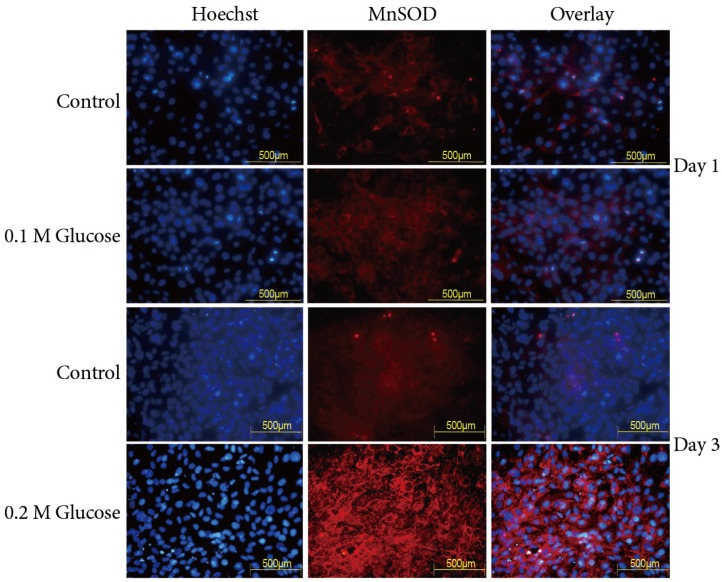
2. Enhanced MnSOD and catalase expressions
The immunofluorescence demonstrated that the two high glucose concentrations enhanced compensatory expression (red color) of MnSOD in notochordal cells at 1 and 3 days, as compared with the control (Fig. 2). Expression of MnSOD (red color) was detected in the nuclei of notochordal cells, whereas expression of MnSOD was observed in not only the nuclei of notochordal cells but also in the cytoplasm around the nuclei. According to results of the Catalase Assay Kit (Cayman Chemical Co.), 0.1M and 0.2M glucose increased catalase expression (mmoL/min/mL) in rat notochordal cells at 1 day (3.2 vs. 4.5, p<0.05; 3.2 vs. 5.2, p<0.05), as compared to the normal control. In addition, 0.1M and 0.2M glucose increased expression of catalase (mmoL/min/mL) in rat notochordal cells at 3 days (5.2 vs. 8.9, p<0.05; 5.2 vs. 11.9, p<0.01), as compared to normal control.
3. Decreased telomerase activity
0.1 M and 0.2 M glucose decreased the mean telomerase activity (arbitrary units) in rat notochordal cells at 1 day (2.5 vs. 2.1, p<0.05; 2.5 vs. 1.4, p<0.01), as compared to the normal control. In addition, 0.1 M and 0.2 M glucose decreased the mean telomerase activity (arbitrary units) in rat notochordal cells at 1 day (1.6 vs. 0.7, p<0.01; 1.6 vs. 0.5, p<0.01), as compared to the normal control.
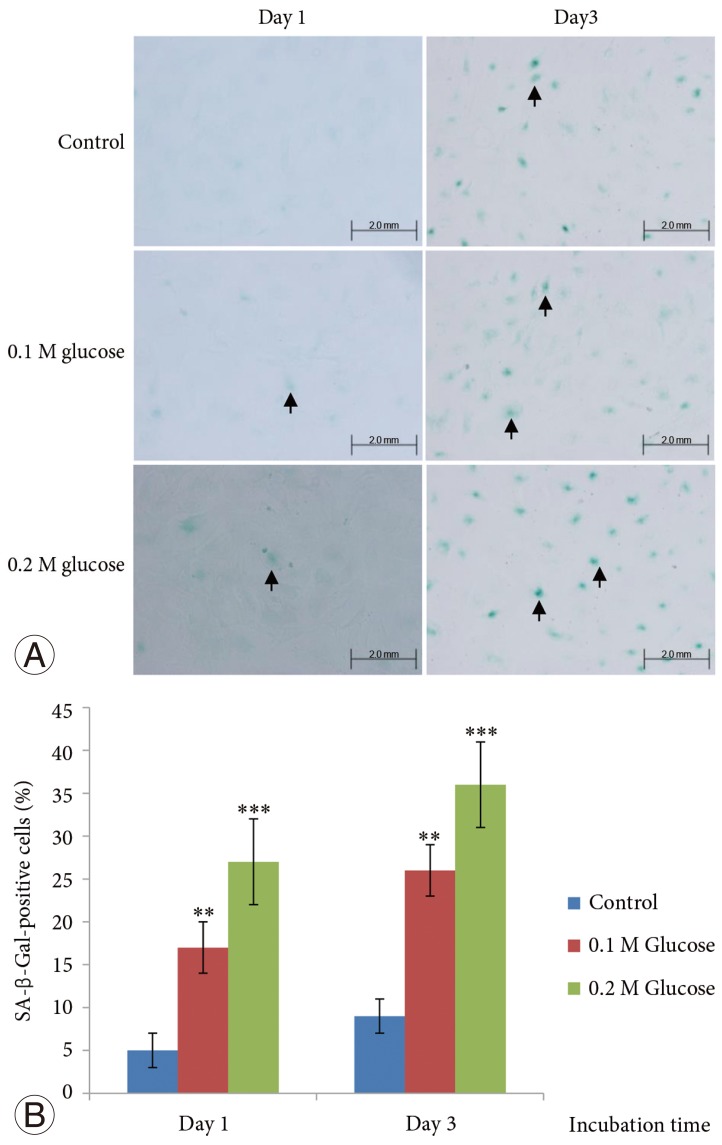
4. Increased stress-induced senescence of NP cells
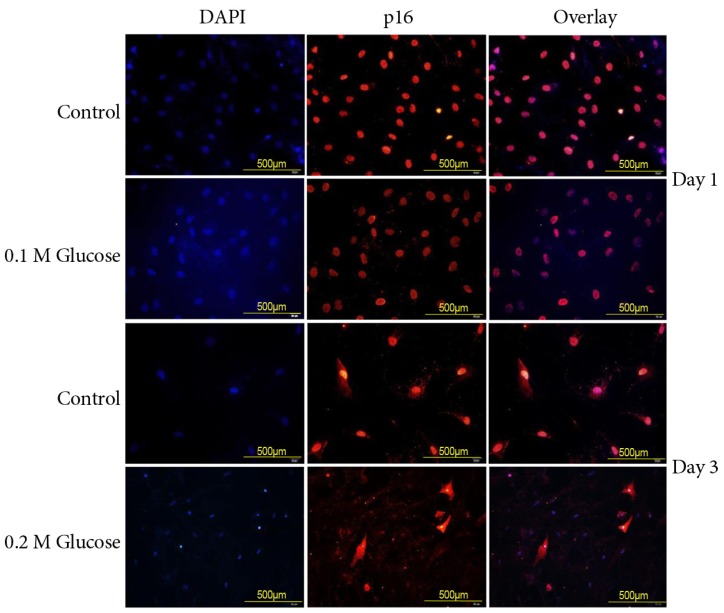
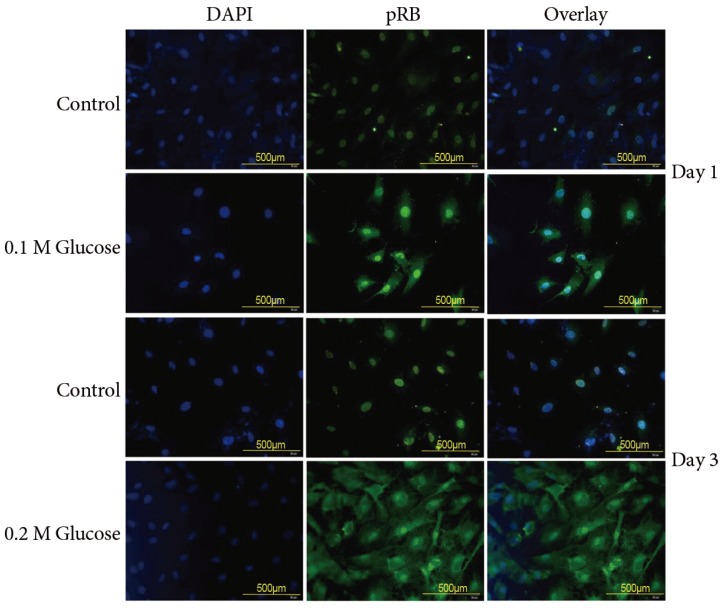
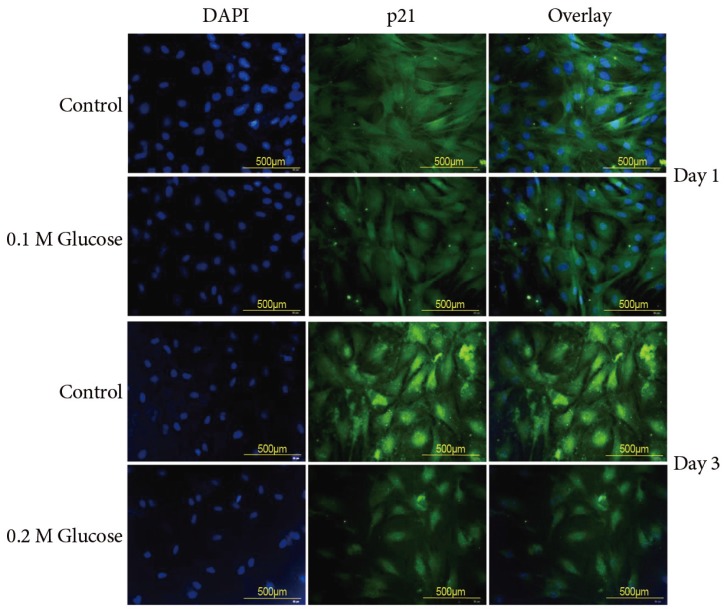
The mean percentage of SA-β-Gal-positive notochordal cells was significantly increased in notochordal cells treated with both high glucose concentrations for 1 and 3 days in a dose- and time-dependent manner, as compared with the control (Fig. 3). Immunofluorescence demonstrated that the two high glucose concentrations enhanced expression of p16 (Fig. 4) and pRB (Fig. 5) proteins in notochordal cells at 1 and 3 days when compared with the controls, respectively. However, immunofluorescence demonstrated that the two high glucose concentrations decreased the expression of p53 (Fig. 6) and p21 (Fig. 7) proteins in notochordal cells at 1 and 3 days when compared with the controls, respectively.
Discussion
The current findings demonstrated that both high glucose concentrations caused mitochondrial damage and consequently increased ROS generation in rat notochordal cells. The occurrence of senescence was significantly increased in notochordal cells treated with high glucose concentrations despite the compensatory expression of antioxidants, such as MnSOD and catalase. Telomerase activity declined in notochordal cells treated with both high glucose concentrations. The expressions of stress-induced senescence (p16-pRB) pathway were enhanced in notochordal cells treated with high glucose concentrations. However, the expressions of proteins related to replicative senescence (p53-p21) were decreased in notochordal cells treated with both concentrations of glucose. These results suggested that high glucose-induced oxidative stress accelerates the stress-induced senescence of notochordal cells through mitochondrial damage. This may result in dysfunction of notochordal cells, leading to accelerated premature disc degeneration. Thus, accelerated stress-induced senescence of notochordal cells could be an emerging risk factor for disc degeneration in young patients with DM. These results indicate the importance of strict blood glucose control in young patients with DM to prevent an excessive generation of oxidative stress and subsequently to prevent or delay premature disc degeneration.
Senescence may have evolved as a mechanism to prevent cells with damaged DNA from being replicated and thus to prevent tumor formation [303132]. Replicative senescence is associated with changes in DNA structure and function including a telomere shortening and decreased telomerase activity [123]. However, senescence appears to be much more complex than a simple cell-cycle arrest occurring after a finite number of cell divisions. Stress-induced senescence can occur due to diverse stimuli including ultraviolet radiation, oxidative damage, activated oncogenes, and chronic inflammation [456]. Oxidative damage to DNA can directly contribute to stress-induced senescence and result in telomere shortening as seen in replicative senescence because the ends of chromosomes are particularly sensitive to oxidative damage [456]. It is well known that damaged mitochondria release harmful ROS into the cytosol [242526272829]. Previous studies reported that antioxidants can protect harmful effects of oxidative stress and extend life-span and reduce age-related changes in tissues such as that of the heart [123]. In the current study, we demonstrated excessive oxidative stress, such as ROS release, through mitochondrial damage in rat notochordal cells treated with high glucose concentrations, as compared to the normal control. However, expression of antioxidants, such as MnSOD and catalase, were also increased in response to both high glucose concentrations, as compared to normal controls. These findings suggested the possibility of a cycle wherein an increased generation of ROS stress leads to compensatory production of antioxidants to neutralize oxidative stress. However, compensatory increased expression of antioxidants could not prevent or reduce the occurrence of premature stress-induced senescence of notochordal cells. We did not comparatively analyze the degree of oxidative stress (ROS) and antioxidants (MnSOD and catalase) in rat notochordal cells treated with high glucose. However, considering the accelerated stress-induced senescence in rat notochordal cells, it is likely that the excessive generation of oxidative stress in rat notochordal cells undergoing high glucose treatment supersedes the protective effect of antioxidants. Further studies are needed to confirm this speculation. Therefore, this result suggested that antioxidants therapy might be ineffective to prevent or delay premature disc degeneration in young patients with DM.
A hallmark of senescence is the expression of SA-β-Gal. However, it is not strictly specific for senescent cells, since non-senescent cells with a high lysosomal content also stain positive in this assay [789]. The current study showed a higher percentage of SA-β-Gal-positive staining indicative of a higher rate of senescence in rat notochordal cells treated with both high glucose concentrations, as compared to normal control. To supplement shortcomings of SA-β-Gal staining, we additionally performed immunofluorescence staining to determine the expressions of proteins, such as p53, p21, pRB, and p16, related to replicative and stress-induced senescence pathways. Immunofluorescence showed that the senescent rat notochordal cells undergoing high glucose treatments significantly expressed p16 and pRB proteins, whereas the expressions of p53 and p21 proteins were significantly decreased, as compared to the normal control. We further showed that high glucose concentrations reduce the telomerase activity of rat notochordal cells. These results indicated that high glucose-induced oxidative stress predominantly accelerates premature stress-induced senescence (p16-pRB) in rat notochordal cells.
Prior to this study, it whether high glucose-induced oxidative stress caused diabetes-related notochordal cells' senescence was unknown. We evaluated the effects of high glucose concentrations on the induction of oxidative stress through mitochondrial damage and senescence in rat notochordal cells. Our study demonstrated that the percentage of senescent notochordal cells increased in proportion to the dose and time of high glucose treatment. This may explain the increased risk of disc degeneration with severity and duration of DM, which results in accelerated premature disc degeneration in young patients with DM. There were some limitations to the current study. Firstly, we did not investigate the telomere length. Telomere shortening is a characteristic finding for cellular senescence. Secondly, in vitro high glucose-induced diabetic model cannot perfectly reflect in vivo aspects of disc degeneration with DM, especially in terms of high glucose concentrations and culture period. Thus, further in vivo studies are needed to clarify these limitations.
Conclusions
The current study demonstrated that high glucose-induced oxidative stress accelerates premature stress-induced senescence of rat notochordal cells despite the compensatory expression of antioxidants. This may result in dysfunction of notochordal cells, leading to accelerated premature disc degeneration. Thus, accelerated stress-induced senescence of notochordal cells could be an emerging risk factor for premature disc degeneration in young patients with DM. Therefore, strict blood glucose control could be important to prevent or to delay premature disc degeneration in young patients with DM.