Introduction
Cellular senescence is a program activated by normal cells in response to various types of stress1. One important mechanism responsible for cellular senescence is progressive telomere shortening and eventual telomere dysfunction that occur due to incomplete DNA replication (an end-replication problem) at the telomeres ('replicative senescence' or 'intrinsic senescence)2-6. This end-replication problem can be resolved by a holoenzyme telomerase, which elongates the telomeric DNA in the 5'-to-3' direction6-11. In the absence of telomerase, or when its expression is very low, the telomeric DNA progressively shortens with each round of cell division12. In addition to replicative senescence, cellular senescence can also be induced in a rapid manner by a number of stresses that are independent of telomere shortening ['stress-induced premature senescence' (SIPS) or 'stress or aberrant signaling-induced senescence' (STASIS)]13,14. Such stresses include oxidative stress, DNA damage, oncogenic activity, and other metabolic perturbations15.
Cellular senescence like apoptosis can be viewed as a powerful tumor-suppressor mechanism that withdraws cells with irreparable DNA damages from the cell cycle16,17. Therefore, the senescence signals, that is, a telomere-based one or a stress-based one, trigger a DNA damage response, and this response shares a common signaling pathway that converges on either or both of the well-established two tumor suppressor proteins, p53 (the p53, p21, pRB pathway) and pRB (the p16, -pRB pathway)1,14,15,18-20. In the p53, p21, pRB pathway, senescence stimuli activate p53, which can then induce senescence by activating pRB through p21, a transcriptional target of p53. This senescence can be reversed upon subsequent inactivation of p53. In the p16-pRB pathway, senescence stimuli induce p16, which activates pRB. Once the pRB pathway is engaged by p16, the senescence cannot be reversed by subsequent inactivation of p53, silencing of p16, or inactivation of pRB18. Although there appears to be overlap between the two pathways, the emerging consensus is that the p53, p21, pRB pathway mediates the senescence that is primarily due to telomere shortening, and the p16, pRB pathway is thought to mediate premature senescence1,14,20. However, a population of growing cells suffers from a combination of physiologic stresses that act simultaneously, and the relative importance of the p53, p21, pRB or p16, pRB pathway for the senescence response may differ depending on the tissue and the species of origin1,20. Once cells have entered senescence, they are arrested in the G1 phase of the cell cycle, and they display a characteristic morphology (vacuolated, flattened cells) and gene expression, including that for markers such as a senescence-associated β-galactosidase (SA-β-gal)21,22.
Degenerative changes in the intervertebral disc (IVD) occur as a natural part of aging23. Gruber et al and Roberts et al recently provided important insights regarding the close link between cellular senescence and disc degeneration, based on the observation that SA-β-gal-positive disc cells increased with advancing disc degeneration and increased in herniated discs24,25. However, the mechanism and signaling pathways involved in the senescence of nucleus pulposus (NP) chondrocytes are unknown. Our present in vivo study demonstrated that, with increasing age and advancing disc degeneration, senescent NP chondrocytes increase or accumulate in the NP. The NP chondrocytes in aging discs exhibited characteristic senescent features, such as increased SA-β-gal expression, shortened telomeres, and decreased telomerase activity. We further show that the telomere-based p53, p21, pRB pathway, rather than the stress-based p16-pRB pathway, plays a more important role in the senescence of NP chondrocytes in in vivo conditions.
Materials and Methods
Twenty-five patients (14 female patients and 11 male patients) who underwent open discectomy for symptomatic herniated NP were included in this study. After thorough removal of the protruded or extruded NP fragments, the NP specimens remaining in the central part of the IVD were pooled, and then they were immediately preserved at -75℃. The NP specimens were grouped according to a grading system for IVD degeneration that was based on the preoperative magnetic resonance images (MRI)26: there were 3 patients with grade II degeneration, 17 patients with grade III degeneration, and 5 patients with grade IV degeneration. In practice, no NP specimens with grade I or grade V degeneration were available because there was no case of NP herniation in any individual with grade I degeneration (normal disc), and there were no remnant NP specimens in patients with grade V degeneration (total collapse of the disc space). The mean patient age was 49 years (range and standard deviation [SD]: 20~75, ± 14 years).
1. Senescence-associated β-galactosidase staining
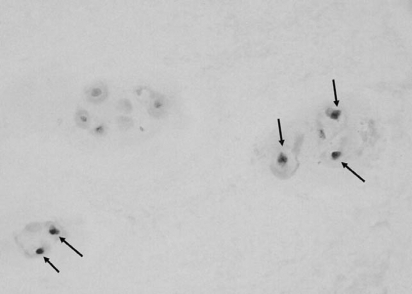
The SA-β-gal activity in the NP chondrocytes was determined using a SA-β-gal staining kit (Cell Signaling Technology). Sections (4 µm thickness) of the frozen NP specimens were placed on slides and fixed with 2% formaldehyde and 0.2% glutaraldehyde in phosphate buffered saline (PBS) for 10 minutes at room temperature. The slides were rinsed with PBS and then incubated overnight at 37℃ with fresh SA-β-gal staining solution containing 40 mM citric acid/sodium phosphate (pH 6.0), 150 mM NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mg/ml of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). After being rinsed with distilled water, the slides were counter-stained with Nuclear Fast Red. All NP chondrocytes and SA-β-gal-positive NP chondrocytes on the whole section were counted (×200), and the percentage of SA-β-gal-positive NP chondrocytes was calculated.
2. Telomere length assay
Genomic DNA was extracted from 40 mg of the NP specimens using the Genomic DNA purification kit (Gentra, Minneapolis, MN, USA). Genomic DNA (2~3 µg) was digested with the restriction enzymes Rsa I and Hinf I at 37℃ for 16 hours and then electrophoresed on 0.6% agarose gels at 50 V for 4 hours. After electrophoresis, the gel was depurinated in 0.25 M HCl for 30 minutes and denatured in 0.4 M NaOH and 1.5 M NaCl for 30 minutes. The DNA was then transferred by capillary transfer onto a positively charged nylon membrane (Hybond-N, Roche; Germany). The membrane was prehybridized in hybridization buffer (TeloTAGGG Telomere Length Assay; Roche, Germany) for 1 hour at 42℃ and then hybridized with a telomere probe in hybridization buffer for 3 hours at 42℃. The membrane was washed twice with washing solution and rinsed with the recommended blocking reagent. The hybridized probe was detected by the chemiluminescence method, according to the manufacturer's recommendation. The membrane was exposed to Hyperfilm (Amersham Pharmacia Biotech, England). The mean telomere length was calculated using the formula: L=Σ (ODi) / Σ (ODi/Li), where ODi is the integrated signal intensity and Li is the DNA length at position i.
3. Telomerase activity
The telomerase activity was analyzed with a TeloTAGGG Telomerase PCR ELISA plus kit (Roche, Germany), according to the manufacturer's instructions. The frozen NP specimens were suspended in 200 µl of ice-cooled lysis reagent and incubated in ice for 30 minutes. The lysate was centrifuged at 16,000×g for 20 minutes at 4℃. The supernatant was carefully removed and transferred into a tube for the TRAP assay. Ten µg of protein extract was used for each assay. Each supernatant was divided into two aliquots. One aliquot was inactivated at 85℃ for 10 minutes and used as a negative control, while the other was used to evaluate the telomerase activity. For each test sample and control, 25 µl of the reaction mixture and 5 µl of the internal standard (IS) were transferred into a tube suitable for PCR amplification. The extended products were amplified by PCR using Taq polymerase, the P1-TS, P2 primers, and nucleotides. After a 30 minute incubation at 25℃ to allow the telomerase-mediated extension of the TS primer, and 5 minutes at 94℃ to inactivate the telomerase, the reaction mixture was subjected to 30 PCR cycles at 94℃ for 30 seconds, 50℃ for 30 seconds, and 72℃ for 90 seconds, then 72℃ for 10 minutes on a thermocycler. Using the ELISA method, the amplified products were immobilized on streptavidin-coated microtiter plates via biotin-streptavidin interaction. The amplicons were then detected by anti-digoxigenin antibodies conjugated to peroxidase. After addition of the peroxidase substrate (3, 3', 5, 5'-tetramethyl benzidine), the amount of TRAP products was determined by measurement of absorbance at 450 nm.
4. Measurement of H2O2 content in the NP tissue
The NP tissues (20 mg) were homogenized in PBS. The homogenate was centrifuged at 15,000 rpm for 15 minutes at 4℃, and the supernatant was collected and stored at -75℃. The H2O2 content was measured using the Quantitative Hydrogen Peroxide Assay Kit (OXIS International, Foster city, CA, USA). This assay is based on the oxidation of ferrous ions (Fe2+) to ferric ions (Fe3+) by H2O2 under acidic conditions. The ferric ion binds with the indicator dye xylenol orange to form a stable colored complex that can be measured at 560 nm.
5. Immunohistochemistry
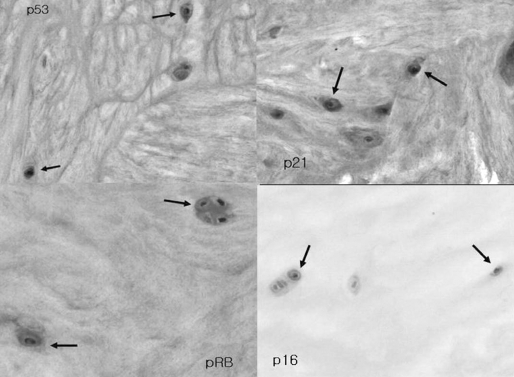
To test the immunohistochemical expression of p53, p21, pRB, and p16INK4 (Phamingen, San Diego, CA, USA), the fresh-frozen specimens stored at -75℃ were embedded in OCT (Tissue-Tek, Sakura Finetek, Torrance, CA, USA). The embedded specimens were cut at -25℃, mounted on lysine-coated glass slides, and then fixed for 10 minutes at 4℃ in 4% paraformaldehyde. The endogenous peroxidase was subsequently blocked by 3% H2O2 for 20 minutes and 0.1% Triton X-100 in PBS for 15 minutes. The cryostat sections were incubated in a humid chamber at 4℃ overnight with the primary antibodies for p16, pRB, p21, and p53. The sections were then exposed to a streptavidin-biotin-peroxidase complex (Histostain Plus kit; Zymed, San Francisco, CA, USA), and the color was developed with 3,3-diaminobenzidine tetrahydrochloride (Lab Vision, Fremont, CA, USA). Mayer's hematoxylin was used for counterstaining.
6. Statistical analysis
The normality of variables (age, percentages of SA-β-gal-positive NP chondrocytes, telomere length, telomerase activity, and H2O2 content) was assessed using the Kolmogorov-Smirnov test. Depending on the normality, correlations among the variables were analyzed using Spearman's or Pearson's test. The differences in the means of the variables among the degeneration grades were analyzed using the Mann-Whitney U test.
Results
1. Senescence-associated β-galactosidase stain
The mean percentage of SA-β-gal-positive NP chondrocytes was 41.82% (range: 15.65~60.80%; SD: ±11.20%) (Fig. 1).
2. Telomere length
The mean telomere length was 14.42 kb (range: 6.60~29.80 kb; SD: ±5.73 kb) (Fig. 2).
3. Telomerase activity
The mean telomerase activity was 6.41 AUs (range: 0.8~22.3 arbitrary units; SD: ±5.37 arbitrary units).
4. H2O2 content in the NP specimens
The H2O2 content in the NP specimens was 0.25 µmol (range: 0.05~0.65 µmol; SD: ±0.14 µmol).
5. Immunohistochemistry
Immunohistochemistry showed that the senescent NP chondrocytes in all 25 specimens expressed p53, p21, and pRB, but only a few NP chondrocytes in two specimens expressed p16 (Fig. 3).
6. Statistical analysis
Among the variables (age, percentage of SA-β-gal-positive NP chondrocytes, telomere length, telomerase activity, and H2O2 content), age and the percentage of SA-β-gal-positive NP chondrocytes showed a normal distribution. The percentages of SA-β-gal-positive NP chondrocytes increased with age (r=0.82, p<0.001), while the telomere length and telomerase activity declined (r=-0.41, p=0.045 and r=-0.52, p=0.008, respectively) (Fig. 4). However, there was no significant correlation between age and the H2O2 content (p=0.18). The NP specimens with grade III (mean±SD: 44.11±9.40%) or IV (mean±SD: 46.02±6.11%) degeneration showed significantly higher percentages of SA-β-gal-positive NP chondrocytes than those with grade II (mean±SD: 21.83±6.01%) degeneration (p=0.01 and p=0.025, respectively).
Discussion
In general, senescent cells become unresponsive to mitogenic stimuli, yet they can remain viable for extended periods of time21. They also express elevated levels of extracellular matrix (ECM)-degrading proteases, collagenases, and matrix metalloproteinase (MMP) family members20,21. In contrast, they express decreased levels of the MMP inhibitor TIMP1 and decreased levels of ECM components, such as elastin, laminin, and several forms of collagen20,21. These alterations indicate a general shift in the phenotype of the senescent cells from matrix-synthesizing to matrix-degrading20,21,27.
Such a shift has also been identified in aging and degenerated IVDs28. The present study showed that the percentages of SA-β-gal-positive NP chondrocytes increases with age. This implies that for NP chondrocytes, like primary cells in other organs of the body, senescence with age is unavoidable22. In addition, the NP specimens with grade III or IV degeneration showed significantly higher percentages of SA-β-gal-positive NP chondrocytes than those with grade II degeneration, which supports the previous finding that the senescent disc cell population increases or accumulates with advancing disc degeneration24,25. Thus, the increase or accumulation of senescent NP chondrocytes with aging may, at least in part, contribute to IVD degeneration, whether it is natural or pathologic.
We further showed that the telomere length and telomerase activity of NP chondrocytes decline with age, while the H2O2 content has no significant correlation with age. In articular cartilage, the chondrocytes were shown to senesce with age via telomere shortening, which was effectively prevented by ectopic telomerase expression29. In contrast, oxidative stress induced by hyperbaric culture conditions caused premature chondrocyte senescence, which was not effectively prevented by ectopic telomerase expression30. Because NP chondrocytes originate from the hyaline cartilage endplate31,32, which is naturally similar to articular cartilage, our results imply that the senescence of NP chondrocytes can be caused by age-dependent telomere shortening and/or age-independent oxidative stress, as occurs in articular chondrocytes. This may explain the increased risk of disc degeneration with age23 and the occurrence of unexpected premature or accelerated disc degeneration in some situations, such as vertebral body fractures33,34.
Although SA-β-gal is a useful senescence marker, its activity is critically dependent on detection conditions. SA-β-gal is also expressed in non-senescent cells that have a high lysosomal content35,36. Multiple markers of senescence are therefore recommended to demonstrate senescence in vivo. For this, we further determined the expressions of p53, p21, pRB, and p16 by immunohistochemistry. Immunohistochemistry showed that the senescent NP chondrocytes in all the specimens expressed p53, p21, and pRB, but only a few NP chondrocytes in two specimens expressed p16. These results indicate that the telomere-based p53, p21, pRB pathway, rather than the stress-based p16, pRB pathway, plays a more important role in the senescence of NP chondrocytes. However, the co-expression of p16, although uncommon, implies that there is a situation in which both pathways are activated simultaneously. Thus, our results suggest that NP chondrocytes in in vivo conditions are subject to a number of physiological stresses that act simultaneously, and the total stress magnitude may determine the appropriate senescence response1.
The results of our study, which addressed disc herniation, may not be applicable to other degenerative disc diseases. In addition, the NP chondrocytes in the herniated NP fragments may senesce via different signaling pathways, or they may undergo apoptosis rather than senescence37,38. To minimize any potential bias related to disc herniation, we consistently obtained NP specimens from the central part of the disc after thorough removal of the protruded or extruded disc fragments. Further studies are required to understand the senescence mechanisms of IVD cells in other degenerative disc diseases.
Although cellular senescence helps an organism by suppressing life-threatening tumorogenesis16,17, it can also be detrimental to the organism by depleting renewable tissues that contain proliferation-competent progenitor or stem cells. In turn, such depletion can compromise the structure and function of tissues, which is a hallmark of aging. Furthermore, senescent cells can persist and acquire altered functions, and so this alters the tissue microenvironments in ways that can promote aging phenotypes20,27,39. The direct relationship between cellular senescence and IVD aging or degeneration is unclear. However, two recent studies24,25, in addition to ours, have provided evidence for the potential role of senescence of disc cells or NP chondrocytes in aging or degenerating IVDs.
In conclusion, the present in vivo study demonstrates that, with increasing age and advancing disc degeneration, senescent NP chondrocytes increase or accumulate in the NP. The senescent NP chondrocytes exhibited characteristic senescent features, such as increased SA-β-gal expression, shortened telomeres, and decreased telomerase activity. Further, we showed that the telomere-based p53, p21, pRB pathway, rather than the stress-based p16, pRB pathway, plays a more important role in the senescence of NP chondrocytes in in vivo conditions, although there is probably a scenario in which both pathways are activated simultaneously. Because the p53, p21, pRB pathway is reversible, prevention or reversal of NP chondrocyte senescence could be a novel mechanism for treating human disc degeneration.