Segmental Spinal Dysgenesis–“Redefined”
Article information
Abstract
Study Design
Retrospective single institutional observational study.
Purpose
Segmental spinal dysgenesis (SSD), a complex spinal dysraphic state caused by notochord malformation disorders, is named after its morphological presentation where a spine segment is dysgenetic, malformed or absent. This study’s objective was to examine and reassess SSD imaging findings and correlate them with an embryological explanation.
Overview of Literature
Scott and his colleagues defined SSD as segmental agenesis or dysgenesis of the lumbar or thoracolumbar vertebrae and underlying spinal cord. Tortori-Donati and his colleagues defined it as a morphologic continuum ranging from hypoplasia to an absent spinal cord segment.
Methods
Fifteen children, whose imaging findings and clinical features were consistent with SSD, were included in the study. Magnetic resonance imaging (MRI) was performed per institutional spine protocol.
Results
Five children (33.3%) presented with a high-ending bulbous cord with no caudal segment, six (40%) presented with a dorsal or lumbar segmental dysgenetic cord with a low-lying, bulky caudal cord but without significant spinal canal narrowing, and four (26.6%) presented with segmental caudal dysgenesis with severe kyphoscoliosis, gibbus deformity, and spinal canal narrowing with a normal distal segment (normal or low-lying).
Conclusions
SSD is a complex spinal anomaly in children requiring clinical-radiological assessment followed by multidisciplinary management based on the extent and severity of the dysgenetic cord and the type of SSD. MRI plays a crucial role in both diagnosing and classifying SSD prior to surgical treatment to prevent further impairment.
Introduction
Segmental spinal dysgenesis (SSD) is a complex congenital spinal anomaly characterized by localized agenesis or dysgenesis of the lumbar or thoracolumbar spine, severe congenital kyphosis or kyphoscoliosis, and focal abnormalities of the spinal cord and nerve roots [1,2]. There is segmental absence or malformation of the spinal cord with a normal distal spinal segment giving the appearance of embryological amnesia [2] (Fig. 1A–C). Characteristically, there is a normal upper spinal cord, a markedly abnormal (either thinned or even indiscernible) affected cord segment devoid of nerve roots, and a bulky, thickened distal cord [1]. The clinical picture of the dysmorphism may vary according to the extent and level of the abnormality, the degree of the resulting kyphosis, and the presence of associated abnormalities. Although Scott et al. [2] defined SSD as an entity involving the lumbar or thoracolumbar spine, Bristol et al. [3] presented case reports of four children, one of whom showed cervicothoracic region involvement. Zana et al. [4] also reported an atypical SSD presentation with multisegmental spinal cord narrowing. Since there is a prevalence of atypical SSD presentations, there is a need for a clear definition of SSD types. Therefore, we attempted to examine and reassess SSD’s radiological features based on its embryological origin.
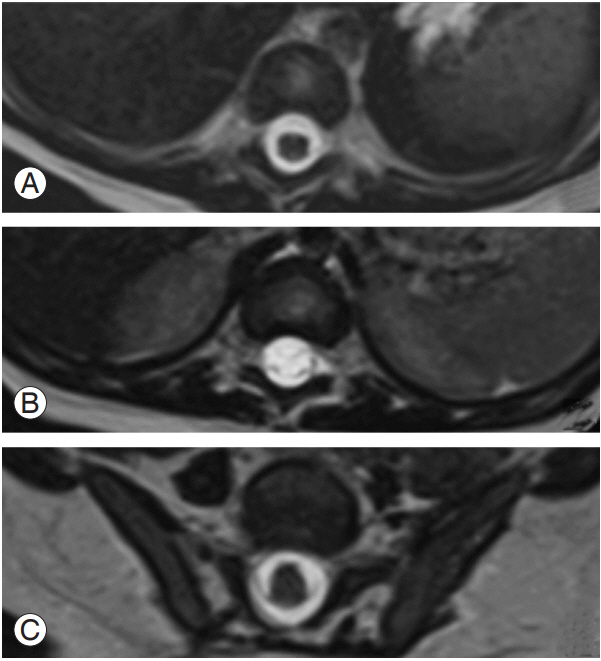
(A–C) Sequential T2 axial sections of the lumbar spine at the D10–D11, L1–L2, and L5–S1 levels. (A) and (C) show the normal cord lying in the spinal canal, whereas (B) shows intersegmental cord absence.
Materials and Methods
1. Demographic data
The present retrospective observational study examines pediatric spine examinations presented at Government Stanley Medical College & Hospital, Chennai-1 between January 2010 and December 2017. Fifteen cases were selected based on the following inclusion criteria: Age <15 years, positive history of congenital paraplegia or paraparesis, presence of segmental spinal cord abnormality, and underlying nerve roots and congenital lower limb abnormalities. Exclusion criteria included postoperative spine cases and scoliosis with no cord abnormality. This study was approved by the independent ethical committee of Government Stanley Medical College & Hospital, Chennai-1 (approval no., 30/2018).
The patients included nine boys and six girls aged between 20 days and 11 years at presentation. All children underwent spinal cord imaging through magnetic resonance imaging (MRI, MR Magnetom Aera, 1.5 T; Siemens, Erlangen, Germany) per institution protocol. The magnetic resonance (MR) spine protocol was as follows: (1) MR myelogram in sagittal and coronal plane; (2) T1, T2, and short T1 inversion recovery images in sagittal sections with a field of view including the entire spine with a slice thickness of 3 mm and slice gap of 0.3 mm; and (3) T1- and T2-weighted axial section of the spine. In all 15 cases, in addition to the whole spine sagittal images, focused axial sections of the malformed cord segment, with the same 3-mm slice thickness and 0.3 mm gap, were imaged for clear visualization of the spinal cord at the level of dysgenesis.
The MR images were systematically analyzed by two experienced radiologists for the level of involvement of dysgenetic cord, status of upper and lower cord with respect to dysgenetic segments, presence of associated vertebral anomalies (both contributory and noncontributory to the deformity of the spine), and for any associated cord lesions or spinal dysraphism. Spinal canal narrowing is considered mild when obliteration of the subarachnoid space is <50% and severe when >50% of the spinal canal is obliterated.
2. Clinical analysis from hospital records
Most of the children clinically presented with kyphoscoliosis, paraparesis or paraplegia, and motor impairment; 11 were paraplegic and four had lower limb weakness (paraparetic) with a maximum power range of 3. Reduced tendon reflexes were described eight patients’ examination histories; and four patients reported absent tendon reflexes. Neurogenic bladder was noted in all cases with associated bilateral hydroureteronephrosis. Anorectal anomalies were seen frequently in approximately 11 patients. A summary of the analysis and results are shown in Tables 1 and 2. Associated visceral organ anomalies included a horseshoe-shaped kidney, present in two children, and dextrocardia in one child. Bilateral dysplastic hips were reported in two children and a unilateral dysplastic hip in one child.

Results of assessment of radiologic findings in segmental spinal dysgenesis

Analysis of imaging findings in SSD and CRS
Results
For the imaging analysis, all 15 patients (100%) presented with vertebral segmentation anomalies. Three children had dorsal cord dysgenesis, two had cervicodorsal level dysgenesis, and three had dorsolumbar dysgenesis. Two children had lumbar cord dysgenesis and five had lumbosacral cord dysgenesis. Although none of the cases showed pure cervical involvement, two children had cervicodorsal interruption of the spinal cord. All children presented with multiple vertebral formation and segmentation anomalies at different levels, irrespective of the dysgenesis level in the cord. Severe scoliosis with gibbus deformity was noted in four cases. In all these patients, the spinal cord was not visible or appeared very thin with acute changes in spinal canal caliber resulting in a wedgeshaped canal at the level of the gibbus.
Dysgenesis is apparent radiologically either as nonvisualization of the cord or as a thin cord. In our study, 11 children were had an absent cord at the level of dysgenesis; three children had a thinned-out and atrophic cord. Mixed presentation of a thin cord, involving few cervicodorsal segments followed by an absent cord for a short segment, was observed in one child, with the distal segment ending at the normal level. Five patients had a high bulbous-ending cord at the lumbar level with no cord below it. The tip of the conus medullaris was lower in eight cases and at the normal level in two cases.
A closed spinal dysraphism was associated with four cases, two of which were spina bifida (posterior spinal defect in the lower lumbar and upper sacral levels without herniation of the intraspinal content), one had a lower cord tethered to the sacrum by a tight filum terminale, and one had a filum terminale lipoma. No evidence of associated open spinal dysraphism was noted. The upper cord exhibited syringomyelia in five cases.
Discussion
According to Tortori-Donati et al. [1] and Scott et al. [2], SSD is characterized by localized deformity of the thoracolumbar or lumbar spine associated with abnormal development of the spinal cord. Bristol et al. [3] described cervicodorsal junction involvement, which was evident in the present study, with involvement in any segment from cervical to sacral region. A literature review synopsis on SSD is shown in Table 3 [1,3-7]. The SSD entity is reported in the literature with multiple conflicting opinions over caudal regression and SSD; therefore, there is a need for a clear definition of SSD. Based on our observations on the different imaging and clinical presentations of SSD, we were able to redefine SSD as a rare, complex, congenital, closed spinal dysraphism which meets the following criteria: (1) presence of congenital paraparesis/paraplegia with lower limb abnormalities; (2) multiple (more than one) formation and segmentation anomalies of the vertebra with or without (kypho)scoliosis; (3) absent or malformed segment of the spinal cord and underlying nerve roots involving any spinal segment from the cervical to sacral region; and (4) visualization of the segment of spinal cord distal to the interrupted cord.
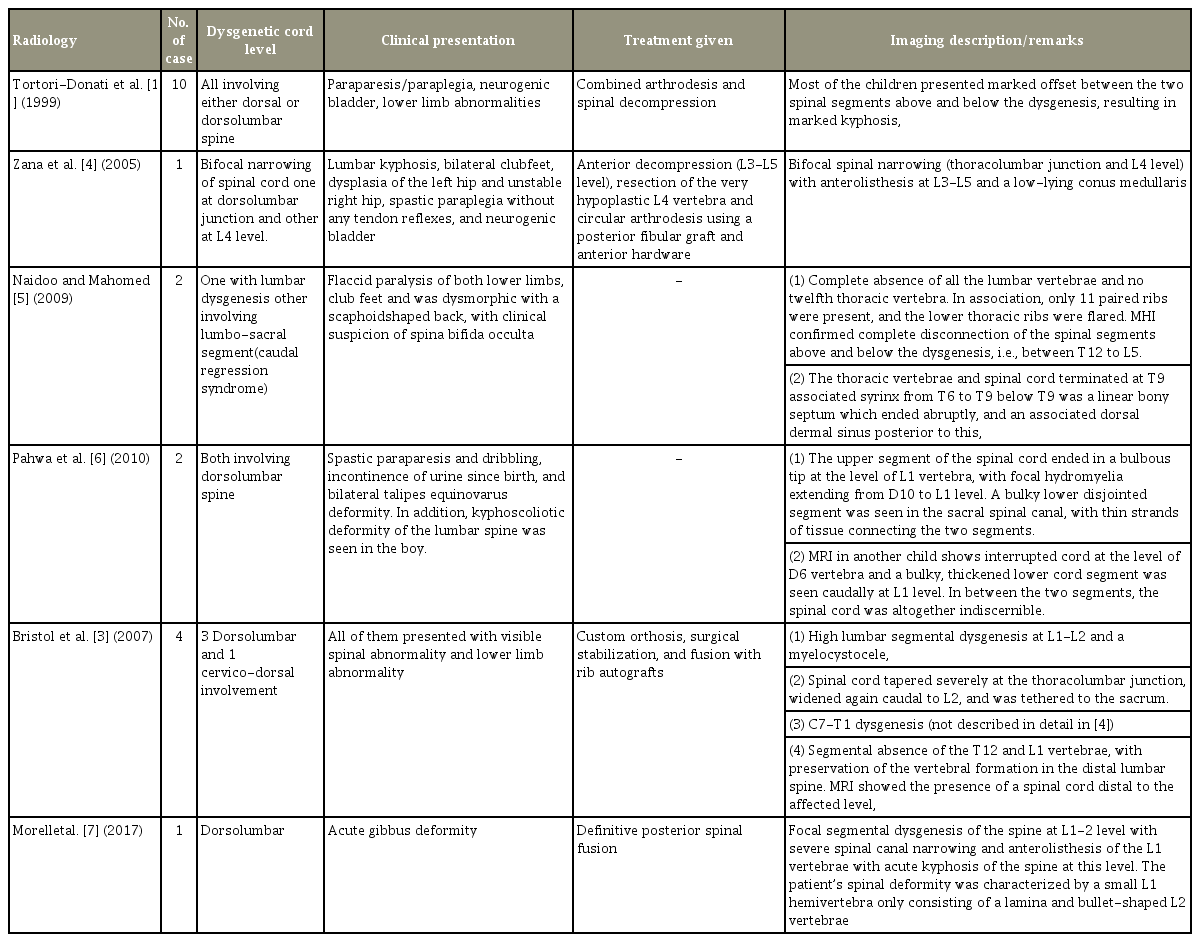
Review of literature on segmental spinal dysgenesis
MRI is a reliable choice for detecting spinal cord abnormalities and should always be performed at presentation. Considering the various imaging presentations and the embryological sequences of SSD, we attempted to classify SSD into two types to assist in further management.
1. Type 1 segmental spinal dysgenesis
Six children presented with mild congenital (kypho)scoliosis. All patients presented without significant spinal canal compromise (Fig. 2A–C). This spinal canal sparing is unique to type 1 SSD (Fig. 3A–C). Focal aplasia or dysplasia of the cord appeared in MRI as segmental absence or as a fibrous, septum-like structure connecting the cranial and caudal ends of the spinal cord. However, the lower cord is almost always bulky and low-lying in type 1 SSD, though it can be normal or low-lying in type 2 SSD.
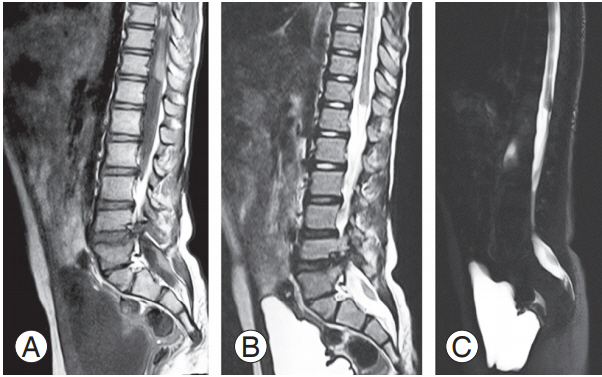
(A) MRI T2 whole spine sagittal section shows the abruptly ending dorsal cord at the D11 vertebral level, with segmental absence of the cord beyond it. (B) A close-up view of the segmental dysgenesis where the low-lying caudal cord is seen separately in the lower spinal canal from L5–S1 to S1–S2. Congenital cartilaginous fusion of the L3, L4 vertebral body is seen with absence of the lower sacrum and coccyx (coccygeal agenesis). (C) Coronal magnetic resonance myelogram shows an absent lumbosacral cord segment with a normal low -lying distal cord segment. Mild narrowing of spinal canal may be noted at the level of L3–L4. MRI, magnetic resonance imaging.
(A, B) T1 and T2 sagittal lumbar spine shows a blunt-ending spinal cord at the D12 vertebra, with conus seen separately in the lower spinal canal (S1–S2 level) without significant spinal canal narrowing (type 1 segmental spinal dysgenesis). Right S2 hemivertebra with mild scoliosis is seen with convexity toward the left (coronal not provided). (C) Magnetic resonance myelogram in the sagittal plane shows the absent cord segment from the D12 to L1 segment. Note the associated neurogenic bladder.
The key process affected in type 1 SSD is gastrulation [1,8], a process by which the bilaminar embryonic disk is converted into a trilaminar disk by the 3rd week of gestation. The ectodermal cells start migrating toward the caudal primitive streak and pass inward at the primitive pit site to fan out or spread between the ectoderm and endoderm to form the mesoderm, which rejoins in the midline to form the notochord (chordamesoderm or axial mesoderm). Whenever this paired analagen fails to fuse in certain areas or remains separate to develop independently, it leads to multiple complex dysraphic states [8].
The chordamesoderm migrating in the ectoderm-endoderm interface has a genetically predetermined destination in its longitudinal axis; therefore, in case of any positioning error in the respected chordamesoderm, apoptosis leads to death or elimination of the malpositioned mesoderm [2]. This results in a hypoplastic or absent cord in different levels based on embryological malpositioning in type 1 SSD.
The notochordal process not only induces the overlying ectoderm to develop into a neural tube, but also induces vertebral body formation through proper induction of the paraxial mesoderm (somites). Therefore, whenever there is a paucity of segmental chorda-mesodermal embryologic elements, it affects development of the spinal column, spinal cord, and nerve roots by altering induction of somite development. A morphologically hypoplastic or absent cord is cephalad to bony abnormality, which is probably due to the metameric relationship between each vertebra and the corresponding spinal cord level.
We defined type 1 SSD as the congenital segmental absence or dysgenesis of the vertebrae, cord, and corresponding nerve roots without significant retrospinal protrusion, gibbus deformity, or spinal canal narrowing due to a chordamesoderm positional error, with its apoptosis leading to primary cord hypoplasia or dysplasia. There is mild to moderate (kypho)scoliosis with a low-lying, bulky distal cord. As there is primary loss of embryogenesis of the spinal cord, surgery has no role in treating type 1 SSD.
The bulky lower cord segment is due to normal elongation of the notochordal process, which occurs by caudal addition of cells [8]. If the derangement occurs in one particular chordamesodermal segment, subsequent migration in the longitudinal axis is not affected. They evade positional apoptosis, leading to an increased number of cells to form the lower lumbar neuroectoderm; this eventually leads to a thicker lower cord segment being formed. In this respect, caudal regression syndrome is embryologically different because of the lower cord’s absence.
Normally, 42–44 pairs of somites are formed with each somite differentiating into a dermatome (skin), a myotome (muscle), and a sclerotome (future cartilage, bone, and ligaments of the spinal column) [9]. In case of any loss of a particular notochordal segment, only the sclerotomal cells are affected without affecting dermatome or myotome cells, as shown in avian embryos [9]. A wide variety of segmentation anomalies of the vertebral column and costal anomalies, particularly in dorsal spine dysgenesis, result without affecting the future skin (dermatome), leading to the nonexistence of open spinal dysraphism in SSD.
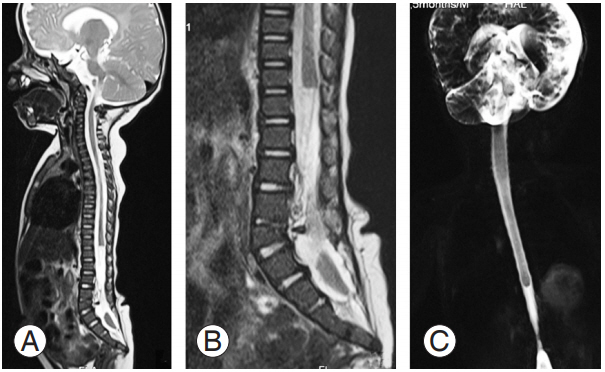
The absence of underlying nerve roots can be explained by apoptotic elimination of the particular somatic neurons, which has been noted in previous studies of vitamin A-deficient quail embryos [10]. In cases in which dysgenesis involves the lower lumbosacral spine, the segmental anomaly level is too caudal for a spinal cord segment to develop below; in these cases, there is an absence of the lower cord caudal to the hypoplastic cord segment (Fig. 4A–C).
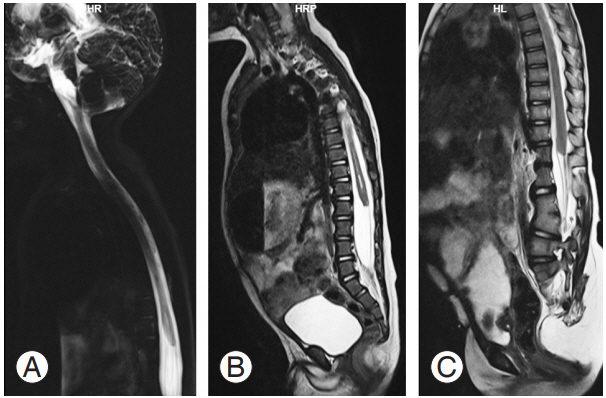
(A) Magnetic resonance sagittal myelogram shows a high bulbous-ending cord without a caudal cord (caudal regression syndrome). (B) T2 sagittal section of entire spine shows the same finding with mild prominence of the central canal in the lower dorsal level. Sacral agenesis, involving the lower sacrum and coccyx, is also seen. Multiple vertebral segmentation fusion anomalies in the cervical and upper dorsal spine with scoliosis and convexity to right are also present (difficult to appreciate in given image). (C) T2 sagittal section of entire spine of another patient, showing high bulbous-ending cord at the upper border of the D12 vertebrae level (distal spinal cord hypoplasia). L1 left hemi vertebrae, fusion of the L3–L4 vertebrae, and sacral agenesis involving the lower sacral vertebrae are seen. Complex lumbosacral vertebral formation and segmental anomaly are noted with mild dorsolumbar scoliosis (convexity to the left).
2. Type 2 segmental spinal dysgenesis
In type 2 SSD, there is congenital segmental absence or malformation of multiple vertebrae, the spinal cord, and its underlying nerve roots to cause severe kyphoscoliosis, gibbus deformity. In contrast to type 1 SSD, there is severe spinal canal narrowing in all patients. The spinal cord at the dysgenetic segment is grossly stretched, compressed, and appears to be thinned-out in segments adjacent to the gibbus apex, with severe spinal canal narrowing (Fig. 5A–C).

(A, B) T2 sagittal section of the spine shows bifocal involvement in both the cervical and dorsolumbar segments. There is cord interruption from the D8–L3 vertebra levels due to herniation of the mid lumbar vertebrae in to the lower thoracic spinal canal, causing severe spinal canal narrowing and gibbus deformity. The conus and lower dorsal cord appear normal. The conus ends at the L–S1 vertebra level, tethered by thick filum terminale lipoma (type 2 SSD). Complete spondyloptosis of C6 over C7 is seen with mild compression of the lower cervical cord. There is syringomyelia of the cord from the D1–D6 vertebral level with focal agenesis/dysgenesis in the lower dorsa lumbar vertebrae. (C) Lumbar spine of another type 2 SSD patient; there is acute kyphosis at the D12 vertebrae level with a small, disc-like D12 vertebral body and non-visualization of the cord from the D9 vertebrae to the upper border of the L1 vertebra. Conus ends at the upper border of the L4 vertebral level. SSD, segmental spinal dysgenesis.
The causal event of type 2 SSD occurs during the formation of spine by somitogenesis (3–6 weeks). Segmentation abnormality and the somite resegmentation results in vertebral anomalies, including hemivertebrae, block vertebrae, butterfly vertebrae, and transitional vertebrae [11]. These anomalies can cause spinal cord compression due to shape alterations, the number of vertebrae, and deformation of the vertebral canal and spinal curvature. This type of spine malformation, which occurs during gestation, is referred to as a congenital vertebral defect.
Spinal cord narrowing and thinning at the gibbus apex level due to vertebral anomalies intra-uterinely can be explained by the two hypotheses: (1) mechanical instability, leading to in-utero spondyloptosis, and cord compression causing chronic ischemia leading to its hypoplasia (dubousset, etc.) and (2) gradual encroachment of the canal by the growth of dysmorphic vertebra. This may be the cause of progressive neurological impairment in type 2 SSD.
Therefore, type 2 SSD is defined as congenital segmental absence or segmental malformation of the vertebrae, spinal cord, and corresponding nerve roots with severe kyphoscoliosis, gibbus deformity, and spinal canal narrowing (obliteration of subarachnoid space >50%) due to abnormal somitogenesis and segmentation anomalies causing secondary cord dysplasia (due to mechanical compression). This abnormal somitogenesis can be congenital. Mutations in the Notch signaling pathway are considered (delta-like 3 and posterior bHLH transcription factor MESP 2) to be one etiological factor [12]. Surgical management, involving complete resection of the dysraphic vertebrae with rib strut grafting, and posterior arthrodesis are recommended to reduce the severity and progressive development of neurological impairment [13].
Although there is evidence of surgical options in the literature, we were not able to predict the precise prognosis of disease as our study subjects did not undergo corrective surgeries.
Conclusions
SSD is a rarely reported, complex spinal dysraphic anomaly in children, for which imaging studies need to be clearly defined and described by a radiologist in order to manage the condition. It usually requires a multidisciplinary management approach based not only on the extent and severity of the dysgenetic cord, but also on the cause (type 1 primary cord hypoplasia or type 2 secondary cord hypoplasia due to dysmorphic vertebra) of the spinal dysgenesis. This article provides a clear definition of SSD and describes newly proposed SSD types to help identify the ideal candidate (type 2 SSD) for further surgical management and reduce progressive neurological impairment due to structural abnormality.
Notes
No potential conflict of interest relevant to this article was reported.
Author contributions
Amarnath Chellathurai: conception & design, analysis of data, critical revision, and administrative support; Balaji Ayyamperumal: critical revision, drafting of manuscript, and analysis of data; Rajakumari Thirumaran: data acquisition, analysis of data, drafting of manuscript, and critical revision; Gopinathan Kathirvelu: analysis of data and data acquisition; Priya Muthaiyan: obtaining funding and supervision; and Sivakumar Kannappan: supervision.