Severe Spinal Injury in Hirayama Disease
Article information
Abstract
Hirayama disease is a rare neurological disorder characterized by an insidious progressive subacute unilateral or bilateral weakness of the hands and forearm muscles leading to a painless amyotrophy. The disease primarily affects young men in the second to third decades of life. It has always been described as a second motor neuron disease, thus sparing the pyramidal and sensitive pathways. It usually has a slow progression course of 3 to 5 years followed by stabilization. Since its initial description by Keyzo Hirayama in 1959, most cases have been reported in Asia, particularly Japan and India, although the disease reportedly has worldwide distribution.
Introduction
Hirayama disease is a rare neurological disorder characterized by an insidious progressive subacute unilateral or bilateral weakness of the hands and forearm muscles leading to a painless amyotrophy. The disease primarily affects young men in the second to third decades of life. It is considered to be a consequence of the dynamic compression of the spinal cord during neck flexion by a detachable posterior dural sac. It has always been described as a second motor neuron disease, sparing the pyramidal and sensitive pathways. It is characterized by unilateral or bilateral upper limb weakness that usually has a slow progression course of 3 to 5 years followed by stabilization [1234].
Since its initial description by Hirayama et al. [1] in 1959, most cases have been reported in Asia, particularly Japan and India, although the disease reportedly has worldwide distribution [34].
Diagnosis is based on the combination of the clinical presentation, neuroimaging and electromyography [345678]. As opposed to cervical myopathy, this disease entity affects only young people, primarily males, with a particular and exclusive weakness distribution of C7-T1 myotomes and the characteristic finding of dorsal dural displacement [8].
Case Report
We described a case of Hirayama disease in a 24-year-old man who was referred to Ramos Mejía Hospital with a 4-year history of slowly progressive asymmetrical weakness and loss of muscle mass in hands and forearms. The patient had no previous medical history and no family history of neuromuscular disease. He is reported to have a maternal great-grandmother who had been born in India.
Physical examination revealed severely wasted hands and forearms. Hands showed significant atrophy of the thenar, hypothenar and interosseous muscles that was more severe in the right side (Fig. 1A). His forearms had a particular affected pattern that involved all muscle groups with spared brachioradialis muscle, resulting in oblique amyotrophy (Fig. 1B).
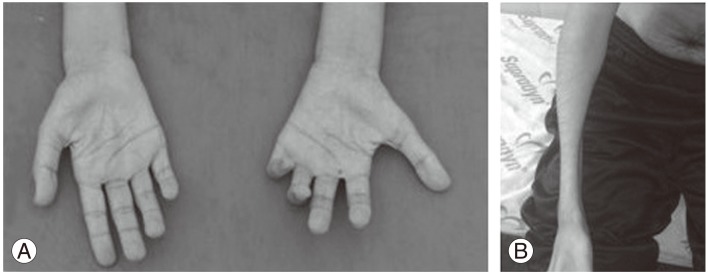
(A) Hands-palmar view. Wasting of the hands with marked thenar and hypothenar atrophy. (B) Right forearm-midpronation/supination view. Wasting of the forearm with sparing of the brachioradialis muscle (oblique amyotrophy).
Muscle strength test showed marked bilateral upper limb deficit including weakness in the wrist extension, wrist flexion, thumb opposition and finger adduction-abduction that was slightly more severe in his right side. There was no weakness in other muscles of the arms or the lower limbs. He had no deficit in any of the sensory modalities. Deep tendon reflexes were brisk, with the exception of left Achilles reflex, which was hyperactive. He had left ankle clonus.
Nerve conduction studies showed low right median and ulnar compound muscle action potential amplitudes. Conduction velocities were within normal range. Median and ulnar antidromic sensory conduction studies were normal. Needle electromyography showed fibrillation potentials and long-duration, high-amplitude, polyphasic motor unit potentials with reduced recruitment in the abductor digiti minimi, extensor indicis, first dorsal interosseous and wrist flexors, indicating the presence of chronic neurogenic pattern with ongoing denervation.
Magnetic resonance imaging (MRI) was performed in a General Electric 1.5 Tesla MRI scanner. Cervical MRI (Fig. 2) at a neutral position showed reduction of spinal cord diameter at C5-C6 level with an elongated central hypertense lesion, symmetrical hyperintense lesions in the anterior horns of the spinal cord (Owl's eyes) and in the axial T2 sequences (Fig. 2A), loss of attachment of the dorsal dural sac to the subjacent lamina (Fig. 2B). Dynamic flexion MRI of the cervical spine showed displacement of the dorsal dural sac from the posterior spinal canal wall with epidural space enlargement with a median anterior dural shift of 3 mm (Fig. 2C). Clinical findings together with electromyographic and neuroimaging studies confirmed the diagnosis of Hirayama disease.
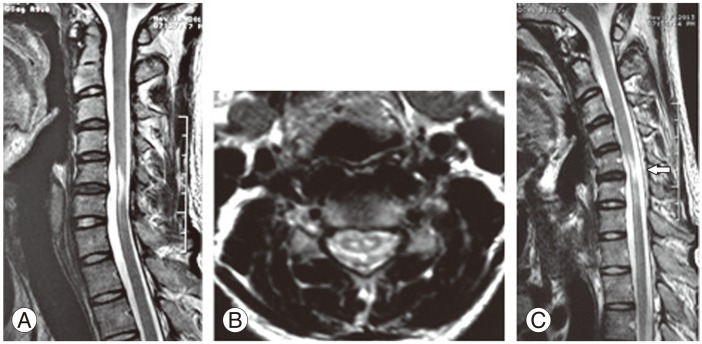
(A) Sagittal fast spin echo T2-weighted images in extension position demonstrates reduced spinal cord diameter at C5-C6 level with a noncompressed elongated central hyperintense lesion. (B) Axial T2-weighted image (433/17 with 20° flip angle) showing loss of attachment between the dural sac and the subjacent laminae with symmetrical hyperintense lesions in the anterior horns of the spinal cord (Owl's eyes sign). (C) Sagittal fast spin echo T2-weighted images in flexion position demonstrate displacement of the dorsal dural sac from the posterior spinal canal wall with enlargement of the epidural space (arrow).
Discussion
Hirayama disease is a motor neuron disorder with a stationary stage after a progressive course. It occurs mainly in males, usually between the ages of 15 and 25 years [5]. Furthermore, it usually affects muscle strength in one upper limb but bilateral cases have also been reported [1234]. Other symptoms like fatigue, cold paresis and tremor have also been reported [2].
It has been described as a cervical flexion myelopathy related to repeated movements of the neck in a tight dural sac [1234567]. The pathogenesis is attributed to a transient forward displacement of the dural space during neck flexion, resulting in anterior horn cell ischemia due to anterior spinal artery compression. It is proposed to occur secondary to an imbalanced length between the vertebral column's growth during puberty and a tightened dural sac [4].
We observed a rare case of bilateral manifestations, distal atrophy and weakness in muscles innervated by C7-C8 and T1 myotomes. The patient had sparing of the brachioradialis muscle (oblique amyotrophy), a characteristic feature of this entity. In contrast to the current descriptions of purely second motor neuron affection, our patient had brisk reflexes, left hyperactive Achilles reflex and a left ankle clonus. To our knowledge, no previous evidence of a patient with pyramidal signs in the lower limbs has been reported. In this case, this is assumed to be a consequence of a severe degree of spinal cord injury, which causes a truly anterior myelomalacia with the subsequent involvement of the corticospinal tract.
Nerve conduction studies revealed variable reduction of median and ulnar compound muscle action potentials. Conduction velocity studies, both motor and sensory, were normal [6]. Needle electromyography findings suggested fibrillation potentials, reduced recruitment, and long-duration motor unit potentials in muscles innervated by the C7-T1 nerve roots associated with anterior horn cell disorders. Hence, all of the electrophysiological signs found in our patient were similar to those previously described.
Conventional MRI studies usually show anterior cord flattening, with various degrees of cord atrophy and pathological high T2 signal secondary to myelomalacia. Findings of loss of attachment in the axial sequences highly supported this diagnosis [348]. Flexion MRI usually shows anterior displacement of the posterior dura mater with an enlarged dorsal epidural space [3]. It corresponds to enlarged veins from negative pressure during the anterior displacement of the dural sac, compression of the anterior epidural veins and a decrease in drainage of jugular veins impeding venous return during neck flexion.
In our case, the patient had all of the radiological signs described above. Interestingly, we found a markedly high signal intensity in sagittal T2 sequences, secondary to anterior horn cell hyperintensity as observed in the axial sequences (Fig. 2A-C). This was an abnormal finding if we consider that in most reported cases, the spinal cord injury is noticeably mild [1]. In the dynamic sequences, there was evidence of a 3 mm-dural detachment as reported in other studies with median anterior dural shift of 3 mm (mean, 2.7 mm; range, 0-7 mm) in patients with this pathology [3]. This suggested that the dimension of the spinal cord lesion is not necessarily proportional to the extent the dura mater detachment.
In conclusion, Hirayama disease should be suspected in young males with a slowly progressive weakness and atrophy of one or both upper limbs. Although rare, it has specific clinical pathologic characteristics, i.e., gender, age, myotome distribution, and MRI findings that can lead to diagnosis.
Notes
Conflict of Interest: No potential conflict of interest relevant to this article was reported.